Presidenta Maite Serrando Querol (desde diciembre de 2019)
Miembros María José Alcaide Martín Paula Argente del Castillo Rodríguez Sandra Gómez Rojas Anna Merino González Ángel Molina Borrás Cristian Morales-Indiano Javier Nieto Moragas Elena Redín Sarasola María Santés Bertó María Sanz de Pedro Xavier Tejedor Ganduxé Eloísa Urrechaga Igartua
Presentación y objetivos
La comisión de Biología Hematológica surge como Grupo de Trabajo en el 2013, con el fin de promover la formación en este campo del laboratorio clínico y divulgar información entre los profesionales del mismo. Cuenta con un equipo de facultativos y residentes dedicados y entregados con pasión al área de la hematología analítica dentro del laboratorio. El área de Hematología en los laboratorios abarca diferentes subáreas relacionadas con los grupos patológicos no neoplásicos y neoplásicos; el objetivo principal de la comisión es exportar el conocimiento de estas diferentes áreas de la hematología a todos los profesionales del laboratorio.
Desde su creación, la comisión ha participado en la organización de numerosas actividades formativas, como cursos presenciales, cursos de casos clínicos ‘online’ o participación en congresos y jornadas divulgativas dentro de las sociedades nacionales de la Medicina de Laboratorio. Además colabora con otros grupos en la realización de documentos divulgativos y tiene como objetivo la elaboración de documentos monográficos y documentos de consenso que ayuden a estandarizar y unificar los criterios entre los diferentes centros y profesionales.
VIII Curso online de Casos Clínicos de Biología Hematológica, Programa 2021-2022.
Documento sobre “Hemoglobinopatías”.
Congreso Nacional del Laboratorio Clínico, noviembre del año 2021, Edición virtual. Curso pre-congreso: Nuevas Tecnologías en el laboratorio de hematología (noviembre 2021).
Documento de “Recomendaciones sobre la determinación de magnitudes de coagulación”, en colaboración con la Comisión de Metrología y Sistemas Analíticos.
Documento sobre “Recuento automatizado de los líquidos biológicos”, en colaboración con la Comisión de Magnitudes Biológicas Relacionadas con la Urgencia Médica.
Documento de consenso sobre “Manejo del paciente con hemocromatosis”, en colaboración con la Comisión de Elementos Traza.
Revisión del documento sobre “Proteínas del metabolismo del hierro. Aplicaciones clínicas”, en colaboración con la Comisión de Proteínas.
Continuar con la divulgación periódica de imágenes de interés en el ámbito de la Citología Hematológica.
Colaboración con la Comisión de Bioquímica de las Enfermedades Inmunológicas y la Comisión de Proteínas. Elaboración del documento sobre la determinación de la triptasa en el laboratorio.
Participación como ponentes de los miembros de la comisión en el “Proyecto European Syllabus Course: Hematology Module” (2021-2022).
Propuesta de Taller de Casos Clínicos en el marco de actividades de la Academia SEQCML.
Memoria de actividades
Organización y/o participación en congresos
Congreso Nacional del Laboratorio Clínico, noviembre de 2018, Bilbao. Curso pre-congreso: Actualización en la interpretación del estudio de anemia. importancia de la serie roja y sus magnitudes relacionadas.
Congreso Nacional del Laboratorio Clínico, noviembre de 2018, Bilbao. Simposio: Contribución del laboratorio clínico al diagnóstico de las leucemias mieloides agudas y papel de las nuevas tecnologías.
Congreso Nacional del Laboratorio Clínico, noviembre de 2017, Málaga. Curso pre-congreso: Interpretación del estudio de líquidos biológicos. Qué puede aportar el laboratorio clínico.
Participación en el VI Simposio Internacional “Laboratorio Clínico y Calidad”, mayo de 2017, Barcelona. Ponencia sobre Novedades en el control de la fase preanalítica en las pruebas de Hemostasia.
Congreso Nacional del Laboratorio Clínico, octubre de 2016, Curso pre-congreso: Papel de la citología de sangre periférica y líquidos biológicos en el diagnóstico desde el laboratorio. Casos clínicos citológicos.
Congreso Nacional del Laboratorio Clínico, octubre de 2016, Desayuno con el experto: La morfología eritrocitaria en el diagnóstico clínico.
Organización y/o participación en jornadas y cursos
Participación en las XV Jornadas del Comité Científico de la SEQCML, marzo de 2017, Madrid. “Detección de leucemias mieloides agudas y neoplasias mieloproliferativas mediante el análisis de sangre periférica”.
Pelcovits A, Niroula R. Acute Myeloid Leukemia: A Review. R I Med J (2013). 2020 Apr 1;103(3):38-40. PMID: 32236160.
Shomali W, Gotlib J. World Health Organization-defined eosinophilic disorders: 2019 update on diagnosis, risk stratification, and management. Am J Hematol. 2019 Oct;94(10):1149-1167. PMID: 31423623.
Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, Orazi A, Tefferi A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018 Feb 9;8(2):15. PMID: 29426921; PMCID: PMC5807384.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. 2016 May 19;127(20):2391-405. Epub 2016 Apr 11.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. 2016 May 19;127(20):2375-90. Epub 2016 Mar 15.
Cazzola M. Introduction to a review series: the 2016 revision of the WHO classification of tumors of hematopoietic and lymphoid tissues. Blood. 2016 May 19;127(20):2361-4. Epub 2016 Apr 11. PMID: 27069255.
Palmer L, Briggs C, McFadden S, Zini G, Burthem J, Rozenberg G et al. ICSH recommendations for the standardization of nomenclature and grading of peripheral blood cell morphological features. Int J Lab Hematol. 2015 Jun;37(3):287-303. Epub 2015 Mar 2.
Recomendaciones para el análisis de las magnitudes biológicas del hemograma (Recomendación, 2021)
Estudio de la ferropenia en el laboratorio clínico (Recomendación, 2018)
Magnitudes biológicas que tiene interés medir de modo urgente (Revisión, 2015)
Imágenes de interés de citología sanguínea
Para los profesionales del Laboratorio Clínico interesados en la citología sanguínea, y con un objetivo docente, en esta sección se publicarán de forma periódica imágenes de frotis de sangre periférica seleccionadas por la comisión, junto a una breve descripción de las alteraciones más significativas.
Esperamos que sea de vuestro agrado.
Paciente varón de 45 años con Eritrodermia. En la morfología de sangre periférica se observan células de estirpe linfoide con citoplasma escaso, núcleo de aspecto cerebriforme denominadas células de Sèzary. Estas células no son exclusivas del Síndrome de Sèzary si no que pueden verse aisladas en sangre periférica de pacientes sin enfermedad (5%) y en otras patologías tanto hematológicas como no hematológicas. Son linfocitos T maduros con fenotipo: CD3+, CD5+ , CD43+ , CD45R0+ , CD45+ , CD4+, CD8-, CD20- y CD30- María Elena Redín Sarasola. Hospital Universitario Donostia
Presencia de un linfocito en sangre periférica de morfología anormal: gran tamaño, elevada relación núcleo-citoplasmática y núcleo de cromatina madura, que muestra pliegues a modo de “circunvoluciones cerebrales”. Anna Merino González. Hospital Clínic de Barcelona.
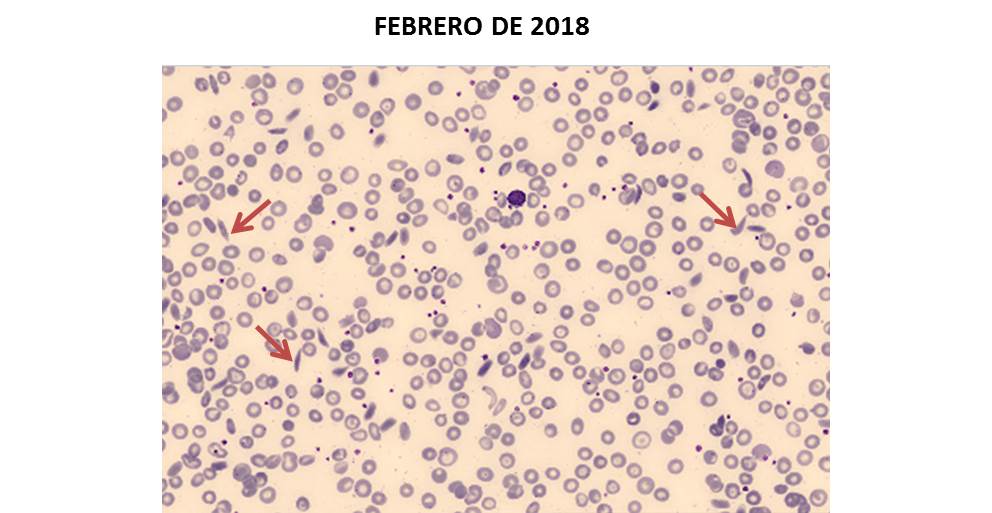
Paciente de 90 años con diagnóstico de SMD presenta anemia sideroblástica. Las anemias sideroblásticas pueden ser adquiridas o congénitas. La anemia sideroblástica adquirida se asocia frecuentemente con un SMD. La síntesis del grupo hemo se ve afectada debido a la incapacidad de incorporar hierro a la protoporfirina IX, induciendo la formación de sideroblastos en anillo en médula ósea. En sangre periférica, destaca la presencia de policromatofília (que indica un aumento del número de reticulocitos) y de eritrocitos con punteado o siderocitos (flechas azules), que contienen gránulos cargados de hierro (cuerpos de Pappenheimer).Eloísa Urrechaga e Imanol Navarro. Hospital Galdakao Usasolo (Bizkaia).
Morfología de la sangre periférica de una paciente de 47 años con pancitopenia y clínica de astenia y fiebre de dos semanas de evolución. Diagnóstico confirmado de leucemia aguda promielocítica. Se aprecia la presencia de células de tamaño mediano, con evidente granulación intensa azurófila y citoplasma color rojo-vinoso. Núcleo de cromatina laxa de difícil observación por la granulación elevada del citoplasma. Dra. Alaitz Vélez de Mendizabal y Dra. Pilar Velárdez López de Ayala. Laboratori Clínic Territorial ICS-IAS, Parc Hospitalari Martí i Julià, Salt (Girona) y Laboratori Hematologia, Institut Català Oncologia (Hospital Dr Josep Trueta, Girona).
Leucemia Mielomonocítica Crónica. varón de 71 años, con monocitosis: 3,5 x103/µL y anemia macrocítica.En el frotis destacan marcados cambios displásicos en serie mieloide: hipogranulación, pseudopelguer, imágenes en espejo y neutrófilos en anillo. Monocitos atípicos con segmentación anómala y cromatina inmadura. Diagnóstico final de Leucemia mielomonocítica crónica tipo 1. Sandra Gómez Rojas. Hospital Universitario 12 de octubre (Madrid).
Presencia de anillo de Cabot y cuerpos de Pappenheimer en el frotis de un paciente con anemia macrocítica y trombopenia, diagnosticado de SMD con sideroblastos en anillo y mutación de SF3B1. La presencia en sangre periférica de distribución irregular de la hemoglobina con punteado basófilo y cuerpos de Pappenheimer sugiere descartar la presencia de sideroblastos en anillo. Sandra Gómez Rojas. Hospital Universitario 12 de octubre (Madrid).
Frotis de sangre periférica de una mujer de 44 años con hepatopatía crónica y con marcadores de anemia hemolítica de larga evolución, con requerimiento de transfusiones frecuentes. Este tipo de anemias suele desarrollarse en el contexto de una cirrosis avanzada y se asocian a un pronóstico desfavorable. Los acantocitos resultan de una alteración estructural en el contenido lipídico de la membrana plasmática, favoreciendo su secuestro y destrucción esplénica. Gonzalo Verdú y Ángel Molina. Hospital Clínic de Barcelona.
Varón de 58 años con adenocarcinoma resistente a tratamiento y con afectación ósea extensa. En sangre periférica presenta pancitopenia (hemoglobina 7 g/dL, plaquetas 15 x 103/µL) en relación a infiltración medular. En la foto observamos elementos de la serie granulocítica en diferentes estadíos intermedios de maduración, blastos y eritroblastos. En relación a la salida de la médula ocupada, podemos encontrar plaquetas o hematíes con alteraciones morfológicas, como los dacriocitos. María Sanz de Pedro y María José Alcaide. Hospital Universitario de La Paz. Madrid.
Paciente mujer de 32 años con leucocitosis (146,4x109/L), neutrofilia (86,4x109/L) y basofília (8.1x109/L). Se acompaña de anemia (hemoglobina 10,5 g/dL) y trombocitosis (1071x109/L). En el frotis se observa mielemia (0,5% promielocitos, 7,5% mielocitos, 7% de metamielocitos) y basofília (5,5%) con algunos elementos degranulados. Se observa algún elemento inmaduro con un recuento inferior al 2%. Algunos neutrófilos presentan la cromatina condensada y otros un núcleo hipersegmentado. Estos hallazgos son compatibles con una leucemia mieloide crónica en fase crónica. Javier Nieto Moragas. Hospital Universitari Doctor Josep Trueta. Girona.
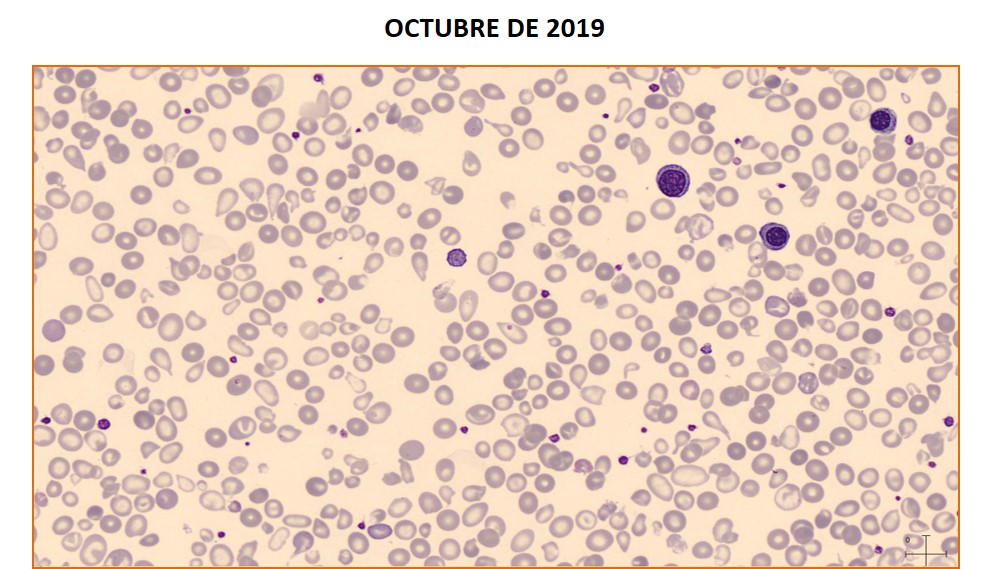
OCTUBRE DE 2022
Paciente de 79 años diagnosticado de neoplasia mieloproliferativa tipo trombocitemia esencial con progresión a mielofibrosis. Acude a urgencias por dolor en hemitórax derecho, febrícula y disnea, objetivándose por radiografía un derrame pleural. En el análisis del líquido pleural se observan células de morfología atípica: tamaño grande, relación núcleo/citoplasma moderada, núcleo de contorno irregular y cromatina laxa con presencia de nucléolo/s visible/s; citoplasma basófilo con presencia de pequeñas vacuolas. El posterior análisis del líquido pleural mediante citometría de flujo confirmó la presencia de un 80% de blastos de estirpe mieloide CD34, CD117 y CD33 positivos. La paciente se diagnosticó de leucemia aguda mieloide (LAM) secundaria a mielofibrosis. Miguel Ángel García Martín y Alicia Martinez-Iribarren. Laboratori Clínic Metropolitana Nord (LCMN). Hospital Universitari Germans Trias i Pujol. Badalona.
AGOSTO DE 2022
Presencia de gametocitos en sangre periférica en una paciente de 42 años, que acude al hospital por fiebre alta, malestar general, cefalea y náuseas a las dos semanas de regresar de un viaje a Guinea. Anna Merino González. Hospital Clínic de Barcelona.
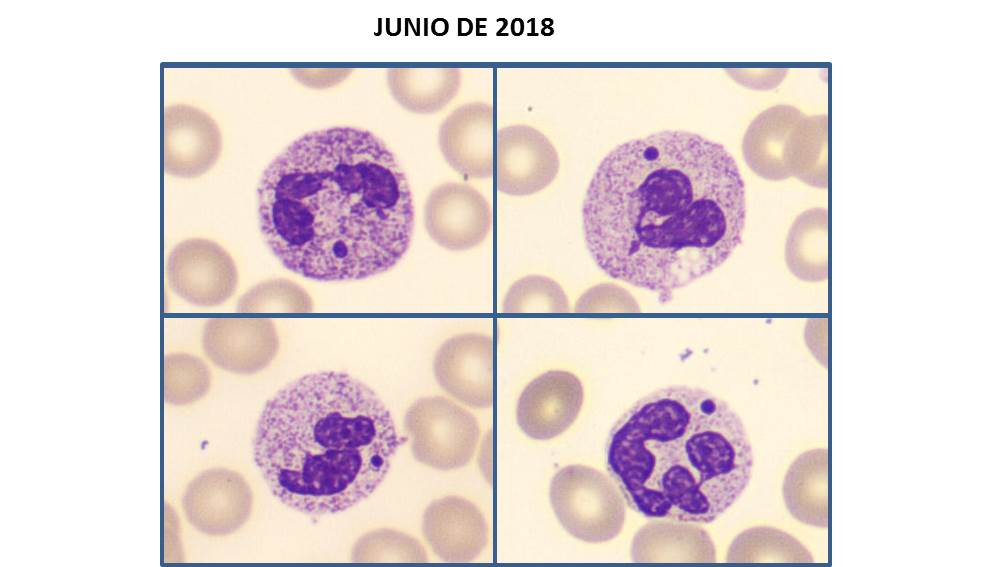
JUNIO DE 2022
Paciente varón de 69 años de edad en cuya analítica de control destaca una linfocitosis de 11,5x109/L junto con alarma de “variant lymphocytes” en el analizador hematológico. En la imagen del frotis de sangre periférica se puede observar la presencia de linfocitos atípicos de aspecto maduro, de tamaño pequeño, relación núcleo/citoplasma elevada, perfil nuclear regular, núcleo con la cromatina madura y citoplasma discretamente basófilo con presencia de pequeñas prolongaciones citoplasmática distribuidas irregularmente. Se amplió estudio en médula ósea objetivándose infiltración nodular y intersticial por neoplasia linfoproliferativa compatible con Linfoma No Hodgkin de la Zona Marginal. Cristian Morales-Indiano y Alicia Martínez Iribarren. Laboratori Clínic Metropolitana Nord (LCMN). Hospital Universitari Germans Trias i Pujol. Badalona.
Imagen de la sangre periférica de un paciente 26 años de edad, que acude a urgencias por fiebre de origen desconocido tras viaje a Perú. Presenta clínica gastrointestinal con aparición de ganglios cervicales bilaterales. En el analizador se aprecia población linfocitaria incrementada con alarma de Atypical lympho que en el microscopio presentan morfología reactiva, hiperbasófilos y con cromatina condensada. La serología fue negativa para CMV y virus EB; positiva para el virus del Dengue. Leire Sáiz Sierra, Maite Serrando Querol. Laboratorio Clínico Territorial ICS-IAS Girona. Parc Hospitalàri Martí i Julià. Salt. Girona.
Paciente de 87 años en seguimiento de síndrome linfoproliferativo crónico tipo LLC-B con deleción en cromosoma 13 (RB1). Diagnosticada hace 8 años e inicio de tratamiento a los 6 años del diagnóstico por la aparición de clínica asociada, con buena respuesta al mismo. En consulta actual presenta una masa axilar de 3 meses de evolución. Ante la sospecha de posible progresión de enfermedad de base, se realiza un TC con contraste y ABMO (aspirado y biopsia de médula ósea), confirmando la misma. Morfológicamente se observan linfocitos de tamaño mediano-grande con núcleos de cromatina laxa, irregulares y algunos con nucléolo prominente (hábito blástico). Estas células siguen expresando algunos marcadores de LLC (CD79a, CD23,CD43) junto con BCL2, MUM1 y un índice de proliferación alto (ki67 > 70%). El cuadro es compatible con una transformación de la LLC a un linfoma no Hodgkin B difuso de célula grande (LNHBDCG). Entre el 2-8 % de los pacientes con LLC pueden evolucionar a un linfoma de alto grado como el LNHBDCG, se denomina Síndrome de Richter, y la supervivencia media es muy baja. María Sanz de Pedro y Mª José Alcaide Martín. Hospital Universitario La Paz. Madrid.
Paciente de 58 años diagnosticado en diciembre de 2015 de mieloma múltiple IgA kappa con tres líneas de tratamiento y progresión cinco años después a leucemia de células plasmáticas. En el análisis hematimétrico se observó un falso aumento de la población de monocitos correspondiente a células plasmáticas de morfología muy atípica. Estas células presentaron un tamaño variable, pérdida de la excentricidad nuclear, a menudo con núcleo multinuclear con uno o más nucléolos visibles y citoplasma de basofília reforzada.Xavier Tejedor Ganduxé. Hospital Universitari Germans Trias i Pujol. Badalona. Barcelona.
Paciente mujer de 49 años con linfocitosis (24,6 x109/L). En el frotis se observa un 92% de linfocitos de morfología pleomórfica. Destacan por un tamaño mediano, relación núcleo-citoplasma moderada, contorno del núcleo irregular i en algunos casos ‘flower-like’. Otros elementos presentan cierta condensación de la cromatina como se observa en la primera foto. Estos hallazgos son compatibles con la leucemia/linfoma T del adulto ligado al virus linfotrópico humano de células T tipo (HTLV-1). Las células en pétalos de flor (‘flower-like’) se pueden observar en la variedad aguda pero también en los portadores asintomáticos de HLTV-1. Algunos linfocitos pueden recordar a la morfología del síndrome de Sezary. Durante el diagnóstico diferencial se deben descartar otros linfomas T cutáneos y otros linfomas T de célula madura. Javier Nieto-Moragas. Laboratorio Clínico Territorial ICS-IAS Girona. Parc Hospitalari Martí i Julià. Salt, Girona.
Presencia de células plasmáticas atípicas en el líquido pleural de una paciente de 49 años que acude a Urgencias por síndrome constitucional y dolor lumbar, evidenciándose un derrame pleural bilateral. En el citospin se observan células de tamaño mediano-grande, con núcleo redondeado de posición excéntrica, citoplasma basófilo y flameado con presencia de arcoplasma. Algunas de ellas son binucleadas. Por citometría de flujo se confirma la presencia de un 25% de células CD138 positivas. A raíz de este hallazgo, la paciente fue diagnosticada de mieloma múltiple IgG lambda. Alicia Martínez-Iribarren y Carla Fernández Prendes. Hospital Universitari Germans Trias i Pujol. Badalona. Barcelona.
Paciente varón de 5 años con leucocitosis (300.000 x 103/µL). En el frotis se aprecian células de aspecto inmaduro, de tamaño grande, relación núcleo citoplasma moderado, núcleo de contorno irregular y citoplasma granulado. Esta morfología sugiere proceso agudo hematológico de probable origen mieloide (aspecto monocitoide). El diagnóstico final fue de Leucemia aguda no linfoide, morfológicamente compatible con LAM M5a de la antigua clasificación de la FAB. María Elena Redín Sarasola. Hospital Universitario Donostia. Donostia, Gipuzkoa.
Extensión de sangre periférica correspondiente a un paciente portador de hemoglobina Köln (Hb Köln). La Hb Köln (HBB Val98Met) es una hemoglobina inestable, de herencia autosómica dominante, con alta afinidad por el oxigeno y que produce de manera variable anemia hemolítica, marcada reticulocitosis y esplenomegalia. A la izquierda (May Grünwald-Giemsa) se puede observar una marcada poiquilocitosis con equinocitos, dianocitos y algún acantocito; debida a la Hb Köln y la presencia de cuerpos de Howell-Jolly (paciente esplenectomizado). La Hb Köln produce cambios de solubilidad en la estructura de la hemoglobina, originando su desnaturalización y precipitación intraeritrocitaria formando cuerpos de Heinz, con hemolisis posterior. La presencia de cuerpos de Heinz se puede observar mediante tinciones vitales como azul de cresilo. En la imagen de la derecha (azul de cresilo) se pueden observar la presencia de abundantes cuerpos de Heinz (flechas) y de reticulocitos (asterisco). Alba Leis Sestayo y Cristian Morales indiano. Hospital Universitari Germans Trias i Pujol. Badalona. Barcelona.
Paciente de varón de 21 años con cuadro clínico compatible con Mononucleosis Infecciosa. En el frotis se observan 47 % de linfocitos grandes con relación núcleo citoplasma baja, citoplasma ligeramente basófilo y en algunos casos se observa indentación por eritrocitos, dibujándose un línea azul. Estos linfocitos se denominan reactivos y aparecen en infecciones víricas principalmente; en ocasiones pueden ser difíciles de diferenciar de los blastos. La serología de Epstein Barr y Citomegalovirus fue negativa y finalmente se pensó en infección por SARS-CoV-2, siendo la PCR positiva. María Elena Redín Sarasola. Hospital Universitario Donostia. Donostia, Gipuzkoa.
Paciente de 62 años portador de VIH que acude por dolor abdominal. En el análisis hematimétrico encontramos anemia junto a trombopenia y se observa un falso aumento de la población de monocitos que corresponde a linfoblastos B (subtipo LAL3) del Linfoma de Burkitt. Estas células presentan un tamaño pequeño o mediano, con un núcleo de perfil redondeado de cromatina laxa e inmadura y con nucléolos visibles. El citoplasma es intensamente basófilo y contiene vacuolas lipídicas. Fernando Calvo Boyero. Hospital 12 de Octubre. Madrid.
Imagen de la sangre periférica de un paciente de 88 años de edad con fiebre y síndrome tóxico. A la exploración física destaca una gran esplenomegalia. El hemograma muestra una linfocitosis absoluta con doble población linfocítica: una predominante de pequeño tamaño, núcleo de cromatina madura y discretamente cuarteada, junto con una segunda constituida por células de gran tamaño, núcleo de cromatina laxa y presencia de nucléolo. Diagnóstico por citometría y citogenética: LINFOMA DEL MANTO.David Cruz. Laboratorio de Hematología. Institut Català d'Oncologia (ICO, Girona). Hospital Universitari Dr. Josep Trueta. Girona.
Presencia de blastos en la morfología de sangre periférica de una paciente mujer de 61 años con diagnóstico de Leucemia Aguda Linfoblástica T (LAL-T). Estas células presentan un tamaño mediano-pequeño, con una elevada relación núcleo-citoplasma. La cromatina de este tipo de células se encuentra moderadamente condensada o laxa, con un nucleolo poco aparente y un perfil nuclear irregular. El citoplasma es escaso y basófilo.Paula Argente del Castillo Rodríguez. Hospital Universitario Son Espases. Palma de Mallorca, Islas Baleares.
Imagen de la sangre periférica de un paciente de 15 años de edad con fiebre y síndrome constitucional de un mes de evolución. Se observó la presencia de precursores inmaduros mieloides (55%, en la imagen) y blastos de morfología mieloide (3%). El hallazgo en una persona joven de leucocitosis, junto a precursores granulocíticos inmaduros (promielocitos y mielocitos), monocitosis y una cifra < 5% de blastos mieloides o monocíticos en sangre periférica es sugestivo de Leucemia Mielomonocítica Juvenil (LMMJ). Orlando Jiménez Romero. Laboratorio Clínico Territorial ICS-IAS Girona, Parc Hospitalàri Martí i Julià. Salt, Girona.
Frotis de sangre periférica teñido con May Grünwald-Giemsa, que corresponde a un paciente de dos años de edad que había ingerido habas. Obsérvese la presencia de numerosos excentrocitos (hematíes con desplazamiento del contenido hemoglobínico a uno o ambos polos). Este hallazgo morfológico nos tiene que hacer pensar en el diagnóstico de un déficit de glucosa-6-fosfato deshidrogenasa (favismo), lo que se demostrará cuantificando la enzima, cuya concentración se encontrará muy disminuída. En estos casos es importante interrogar al paciente sobre la ingesta de determinados alimentos (habas) o fármacos oxidantes. Elena de Rafael González. Hospital Virgen de la salud, Complejo Hospitalario Universitario de Toledo.
Paciente varón de 70 años con diagnóstico de Leucemia Mieloide Aguda Megacarioblástica (LMA M7 de la clasificación de la FAB). En sangre periférica (SP) presenta un 70% de blastos. En la/s imagen/es se observan blastos de linaje megacariocítico, de tamaño mediano-grande, con relación núcleo:citoplasma elevada, núcleo redondeado y mayoritariamente regular con cromatina algo más condensada que otro tipo de blastos y, en ocasiones, con nucléolo/s más o menos evidente/s. En cuanto al citoplasma, éste es basófilo y agranular, y presenta protusiones (“blebs”), proyecciones o pseudópodos. En la SP de estos pacientes pueden observarse además de los blastos, micromegacariocitos, núcleos desnudos de megacariocitos circulantes y plaquetas en general dismórficas. La LMA Megacarioblástica puede presentarse tanto en adultos como en niños. Es una entidad poco común y supone <5 % de los casos de LMA. María Sanz de Pedro, M.ª José Alcaide Martín. Hospital Universitario La Paz, Madrid.
Promielocitos atípicos con núcleo bilobulado o “signo del hachazo” y el de la parte superior presenta astillas. Ante la presencia de estas células hay que sospechar leucemia aguda promielocítica M3, la cual cursa habitualmente con alteraciones de la coagulación (CID) y constituye una urgencia médica. María Helena Redín Sarasola. Hospital Universitario Donostia. Donostia, Gipuzkoa.
Beta Talasemia Mayor. Anisocitosis con marcada poiquilocitosis y policromasia; se aprecian formas eritrocitarias dismórficas entre las que destacan abundantes dacriocitos, algún dianocito y esquistocitos. Se observan eritroblastos, células de aparición frecuente en este tipo de hemoglobinopatía.Maite Serrando Querol. Hospital Universitari Doctor Josep Trueta. Girona.
Presencia de linfocitos anormales en un Síndrome de Sézary (variante leucémica de la micosis fungoide, un linfoma cutáneo de células T maduras). Las células de Sézary pueden presentar dos variantes morfológicas; la variante grande cuyo tamaño es aproximadamente el de un monocito y la variante pequeña (o células de Lutzner) similar al tamaño de un linfocito y morfológicamente más difíciles de reconocer.
Las células de Sézary se caracterizan morfológicamente por presentar una relación núcleo-citoplasma elevada, una cromatina madura sin presencia de nucléolos y un perfil nuclear irregular caracterizado por mostrar evidentes surcos o pliegues superponibles que recuerdan a las circunvoluciones cerebrales y que le confieren un aspecto cerebriforme. En algunas ocasiones pueden presentar pequeñas vacuolas citoplasmáticas. Cristian Morales-Indiano y Carla Fernández Prendes. Hospital Universitari Germans Trias i Pujol. Badalona. Barcelona.
Cristales de hemoglobina en el frotis de sangre periférica. La imagen de la izquierda corresponde a un paciente con hemoglobinopatía doble heterocigota SC. Esta patología se caracteriza por presentar escasos drepanocitos y abundantes dianocitos. En algunos casos pueden encontrarse con cierta frecuencia cristales de hemoglobina SC (flechas), formando agregados que deforman el hematíe. Esto da lugar a poiquilocitos de morfología muy variada en comparación a los cristales que podemos encontrar en la hemoglobinopatía C homocigota (derecha) que muestran una morfología más uniforme que generalmente recuerda a la forma del bacilo. Ángel Molina Borrás. Hospital Clínic. Barcelona.
Tripanosomiasis africana diagnosticada en la revisión del frotis de sangre periférica en una paciente de 49 años, que acude al hospital por fiebre, fatiga y dolores articulares a las dos semanas de regresar de un viaje a Tanzania. Anna Merino González. Hospital Clínic. Barcelona.
Linfocito con incrustaciones en su citoplasma en forma de cristales que corresponden a Inmunoglobulinas. Corresponde a un linfocito B de un paciente con LLC-B (Leucemia linfoide crónica de tipo B). María Helena Redín Sarasola. Hospital Universitario Donostia. Donostia, Gipuzkoa.
Leucemia mieloide aguda con mutación nucleofosmina (NPM+). Obsérvese la “huella digital” o también denominada “cup like” como una depresión a nivel del núcleo de las células blásticas. Anna Merino González. Hospital Clínic. Barcelona.
Paciente varón de 65 años con pancitopenia (recuento de leucocitos: 2,8 x109/L, Hemoglobina: 124 g/L y recuento de plaquetas: 60 x109/L). Ante una pancitopenia el diagnóstico diferencial morfológico incluye fundamentalmente: a) leucemia aguda, b) tricoleucemia y c) anemia aplásica. En estos casos las células diana (a buscar) serían principalmente blastos o tricoleucocitos. En nuestro caso el frotis de sangre periférica analizado mediante el Cellavision DM 1200 reveló la presencia de un 2 % de tricoleucocitos. La presencia de tricoleucocitos en sangre periférica es patognomónica de tricoleucemia. Laura Bigorra López e Iciar Larriba Alegría. Core Hematología. Synlab Diagnósticos Globales. Esplugues de Llobregat. Barcelona.
Parasitación eritrocitaria por Plasmodium ovale. Ante una sospecha de infección por Plasmodium vivax/ovale, uno de los criterios que sirven de ayuda para diferenciarlos es el número de merozoítos por esquizonte. En este caso pueden distinguirse 7-8 merozoítos/esquizonte característico de P. ovale, mientras que en la infección por P. vivax pueden observarse entre 14-32 merozoitos por esquizonte. Ángel Molina Borrás. Hospital Clínic. Barcelona.
Presencia de inclusiones Howell-Jolly Body-Like en el citoplasma de neutrófilos. Estas inclusiones representan fragmentos nucleares de origen apoptótico. Se pueden observar en pacientes inmunodeprimidos en tratamiento con quimioterapia o con antivirales análogos de nucleósidos (aciclovir, ganciclovir, etc…), responsables de la fragmentación nuclear. Es importante reconocer este tipo de inclusiones y poder diferenciarlas de otras también presentes en el citoplasma de los neutrófilos. Cristian Morales-Indiano y Javier Nieto Moragas. Hospital Universitari Germans Trias i Pujol. Badalona. Barcelona.
Frotis de sangre periférica correspondiente a una mujer de 42 años fumadora. Destaca la presencia de linfocitos binucleados o con el núcleo en espejo característicos de una Linfocitosis B Policlonal Persistente (LBPP). Un aumento de la IgM con el resto de inmunoglobulinas normales o disminuidas junto un proteinograma sin alteraciones respaldan la orientación diagnóstica para una LBPP. Cristian Morales-Indiano y Javier Nieto Moragas. Hospital Universitari Germans Trias i Pujol. Badalona. Barcelona.
Hematíes en forma de guadaña o hematíes falciformes. Corresponde a un paciente con una hemoglobinopatía, hemoglobina S conocida como Drepanocitosis o Anemia de células falciformes. María Helena Redín Sarasola. Hospital Universitario Donostia. Donostia, Gipuzkoa.
Frotis de sangre periférica correspondiente a una paciente mujer de 29 años con infección VIH y macrotrombocitopenia (26x109/L) en ausencia de hemorragias significativas e inclusiones basófilas alargadas y triangulares en la periferia de los neutrófilos: Anomalía de May-Hegglin, alteración autosómica dominante debida a una mutación en el gen MYH9. Anna Merino González. Hospital Clínic. Barcelona.
La SEQCML únicamente utiliza cookies técnicas, de personalización y de análisis, propias y de terceros, que en ningún caso tratan datos de carácter personal ni captan hábitos de navegación para fines publicitarios. Si continua navegando, consideramos que acepta nuestra política de cookies.